皮肌炎(dermatomyositis,DM)是一种皮肤肌肉炎症,以模糊的前驱症状、皮炎和肌肉炎症与变性为特征。一般情况下皮肌炎应包括皮肤和肌肉两方面病变,但可表现为单一病变,如无皮肤改变仅有肌肉受累者称为多发性肌炎,而无肌肉改变仅有皮肤特征性损害者称为无肌病性皮肌炎。女性患者为男性的2倍,黑人患者为白人的4倍;有两个发病高峰,低峰见于儿童,高峰见于成人,后者年龄在40~65岁之间。以皮肤和肌肉病变为主,但两者并不平行,即有时以皮肤病变为显著,有时以肌肉病变为显著。有的患者合并恶性肿瘤,可与其他结缔组织病重叠。
1、全身症状 患者多为缓慢起病,少数急性发病;早期常为全身乏力,有间断发热、不适、厌食、关节痛和显著体重减轻的表现,继而出现皮肤疹和肌肉疼痛。
2、皮肤表现
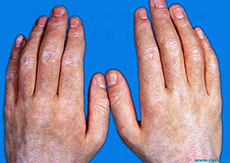
本病通常以颜面和眼睑的红斑、肿胀开始,有时扩展到其他部位。双眼睑常首先受累,呈肿胀和紫红色,可有疼痛和触痛,这是由于眼轮匝肌受累的结果,眼睑上有微细的毛细血管扩张。其他皮肤改变包括红斑性或荨麻疹性斑片和斑块,见于面颊部和四肢,也可有坚实性、轻微凹陷性水肿,特别是在肩胛带区、上臂和颈部;头皮受累以萎缩、红斑脱屑性斑块为特征,有时发生脱发;光敏性常见;在某些患者瘙痒可能较严重。该期可持续数月,继而出现典型的更广泛的皮肤损害,从颜面扩展到颈、胸、肩、臂和其他部位,特征性皮损区包括颈部和上胸部的“V”型,上背、颈和肩部的“围巾“型。一种特征性紫红色脱屑性皮损出现于指节、膝和肘的伸侧,称为Gottron征;在指节上有一些平顶、紫红色的丘疹,称为Gottron丘疹,被认为是皮肌炎最有疾病特征的皮损(图26-5);指尖周围、拇指侧面、手指和偶尔伴手掌等处出现角化过度、脱屑、皲裂和过度色素沉着,这种手部病变称为“技工手”;甲皱褶近端可见毛细血管扩张引起红斑性线状或点状改变,也可形成腊肠形毛细血管袢,类似于硬皮病;雷诺现象和躯体多毛症较少见;粘蛋白局部沉着类似SLE。疾病缓解时可发生过度色素沉着,类似于艾迪生病,皮肤出现青铜色改变;面部和上胸的毛细血管扩张和红斑性皮损可持续数月甚至数年,伴周期性加剧,最后变成棕色色素沉着,这种色素沉着可伴有色素减退、萎缩和毛细血管扩张,最终发展成异色性皮病;偶尔形成大片的持续性溃疡,在屈侧部位和驱体受压点容易发生;皮肤和肌肉钙质沉着见于50%的儿童皮肌炎,而成人则比较少见,与疾病活动期的长短和严重程度有关,主要发生于躯体上半部、肩胛带区、肘部和手部。
3、肌肉改变
严重患者早期可出现严重的肌无力伴急性肿胀和疼痛,对称发生,最常累及肩胛带,有时累及骨盆区和手部,患者常诉双臂不能上举,不能梳头,站立时小腿疼痛,不能上楼梯,吞咽、说话甚至呼吸都感到困难,这是由于疾病早期各部位的肌群受累之故;后期出现萎缩和关节炎改变,导致关节强直;心脏受累伴心衰可见于疾病晚期。
有些患者具有皮肌炎的典型皮肤表现,但无肌肉受累称为无肌病性皮肌炎或无肌炎性皮肌炎,但有肌肉炎症存在而无症状者也较常见,因此除详尽的物理检查以外,还应测定各种肌酶、肌电图和磁共振显像等检查,以便早期发现。
4、伴发疾病
硬皮病改变是最常见的并发症,这种情况称为“硬化性皮肌炎”,在该亚群中有抗Ku抗体和抗PM/sc1抗体;混合结缔组织病和少见的类风湿性关节炎、红斑狼疮及Sjǒgren综合征也可能相伴发生;还可与间质性肺病伴发,患者有抗Jo-1抗体和其他抗合成酶抗体如抗PL-7抗体、抗PL-12抗体、抗DJ抗体和抗EJ抗体。
5、肿瘤与皮肌炎
成人恶性肿瘤常与皮肌炎伴随发生,最常见于50~60岁以上患者,女性比男性更常见,因此在成人皮肌炎患者中(尤其是40岁以上者)根据组织学检查、物理检查和常规实验室检查对恶性肿瘤进行早期发现是必要的。
6、儿童皮肌炎
儿童皮肌炎有些特点与成人型不同,有两种变异型,较常见的是Brunsting型,其病程慢,有进行性肌无力、钙沉着症以及对糖皮质激素治疗敏感,钙沉着症可能发生于皮下和肢端(肘、膝、指),如成人中所见,或为经典型,累及肌间筋膜层;第二型即Banker型,其特点是肌肉和胃肠道的血管炎,迅速发作,严重肌无力,对糖皮质激素治疗不敏感,直至死亡,此型少见。内脏恶性肿瘤偶尔见于儿童皮肌炎患者。